

WASHINGTON, D.C., Feb. 24, 2021 — U.S. national flags fly at half-staff at the Washington Monument … [+]
Random variation is an essential component of all living things. It drives diversity, and it is why there are so many different species. Viruses are no exception. Most viruses are experts at changing genomes to adapt to their environment. We now have evidence that the virus that causes Covid, SARS-CoV-2, not only changes but changes in ways that are significant. This is the twenty-second part of a series of articles on how the virus changes and what that means for humanity. Read the rest: part one, part two, part three, part four, part five, part six, part seven, part eight, part nine, part ten, part eleven, part twelve, part thirteen, part fourteen, part fifteen, part sixteen, part seventeen, part eighteen, part nineteen, part twenty, and part twenty-one.
The variants have caused us to reevaluate our strategies of control for the Covid-19 pandemic. Variants have now popped up from many parts of the world. They confer new and dangerous properties including increased transmission, the ability to evade the immune response elicited from previous infection or vaccination, and increased virulence and infection, especially in children. It is in this context that we describe a Washington DC variant (B.1.189) first identified from an outbreak in pediatric patients. B.1.189 differs from many other variants in that it carries a single, rather than multiple, mutation in the spike protein. Nonetheless, this single mutation appears to alter the virus properties in significant ways
Variant B.1.189, detailed in a study by the Children’s National Hospital in Washington DC, was first identified in mid-October in an infected newborn displaying more aggressive symptoms than most other infected children. Not only was the newborn feverish, but when the doctors measured the viral load, it was over 50,000 times the median of other pediatric patients. This realization prompted greater genome sequencing of young patients in Children’s National Hospital, leading to several more cases of the variant identified.
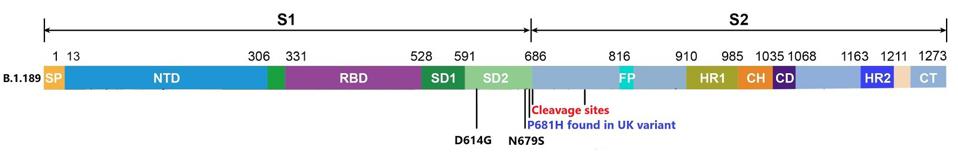
The variant’s mutational makeup is far simpler than most emerging SARS-CoV-2 strains. Aside from D614G, which is included in nearly all circulated viruses today, B.1.189 only carries one additional mutation to the spike protein at position 679—N679S. This mutation has not been previously identified in variants of concern, however, the GISAID database contains several hundred instances of N679K.
Mutations are point 679 are found in two distinct clades. In tracing back the origin, it seems the first appearance of this mutation in the US occurred in Texas, but most recently in the DC area in pediatric cases. The importance of 679 in providing an advantage to the virus is also attested to by detection of the same mutation in an independent lineage in East Asia as well.
MORE FOR YOU
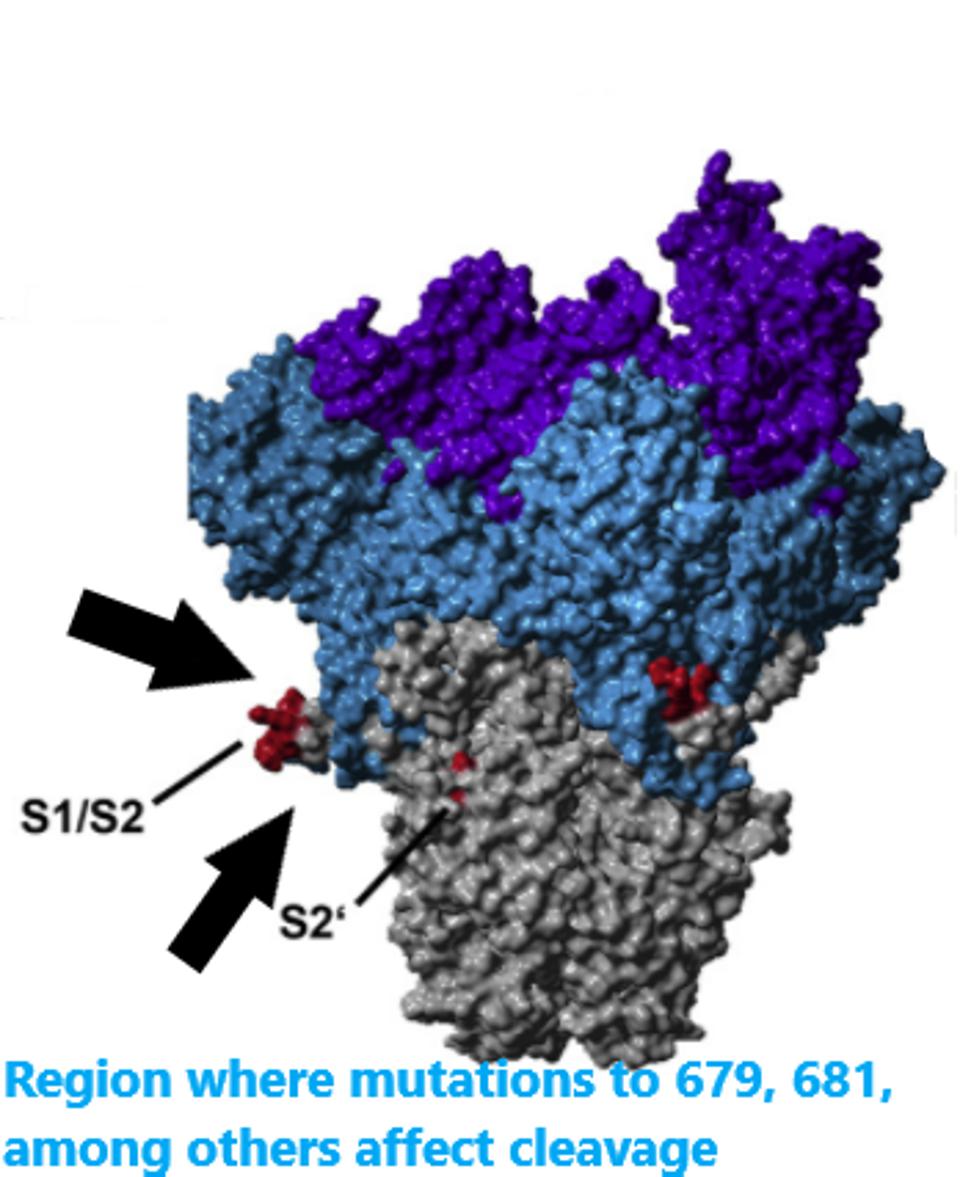
The N679S mutation is located in a critical area for determining the infectivity of the virus particle. For SARS-CoV-2 to affect the cell, two cleavage events are required. One at the furin cleavage site and the other just before the fusion peptide. These cleavage events are the final steps of activation prior to the merger of the virus and the cell.
The researchers speculate that N679S is close to a cleavage site around amino acid 681. The infectivity is increased by these cleavage sites. We propose that the mutation increases the infectivity of the virus by further facilitating the effect. Increasing the rate of cleavage may benefit the virus in two ways: increasing its ability to be transmitted and increasing the efficiency of replication once the infection has been initiated. N679K may also increase infectivity in other variants around the world.
Mutations to the spike protein
Location of the cleavage site and nearby mutations
We also note that the B.1.1.7 variant now circulating widely around the world, which is known to have a substantial increase in transmission, infectivity, and disease potential, is located in the same region and is postulated to have similar activity. It is also notable that B.1.1.7 is recorded to infect and sicken children at a higher rate than strains without these mutations.
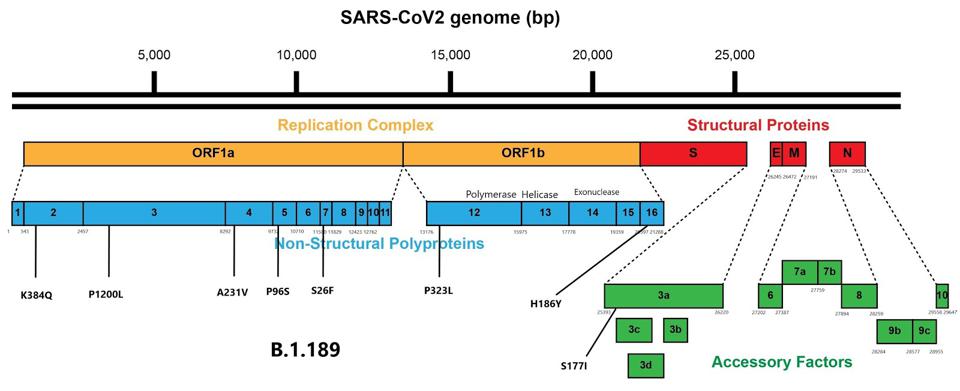
Additionally, B.1.189 carries mutations to several points in ORF1ab and ORF3a. These mutations are commonplace in variants of concern and further enhance replication and virulence. Among these are P1200L in non-structural protein (NSP) 3, which is a transmembrane protein, and a mutation to this mechanism could increase immune escape. Other mutations to transmembrane proteins that could increase immune escape are A231V in NSP4 and P96S in NSP6. S26F in NSP7 and P323L in NSP12 both are involved in transcription and could increase replication competency.
Mutations external to the spike protein
Variant B.1.189’s discovery in children mirrors a rise in SARS-CoV-2 cases among children in other areas of the world. Recent data shows a surge in both Italy and Israel youth cases, the latter experiencing more positive cases for children and teens than in any other month of the pandemic.
The researchers conclude by championing recent genome sequencing efforts. With the rise of the United Kingdom and South Africa variants, the scientific community and political bodies are becoming keenly aware of the threat that new variants represent. While cases have been on a recent decline, in many countries, over the past few weeks those declinations are plateauing. In some cases, such as in the United States, we have seen a slight uptick. Whether that is due to the increased spread of the variants or relaxation in mitigation measures, time will tell.